TransAtlasDB
An Informatics system for Transcriptome Analysis Data
TransAtlasDB Sample Format
This section discusses TransAtlasDB input data format and output formats.
INPUT FORMAT
TransAtlasDB accepts input data from the different software required for differential expression analysis.
The current prototype accepts outputs from the Tuxedo Suite - TopHat and Cufflinks.
- Sample Information.
The Sample information, or samples metadata, is the reference point of the corresponding results from RNAseq data and therefore important for data archival and retrieval of the various transcriptome analysis. TransAtlasDB preferably accepts the sample information through the FAANG sample submission spreadsheet template to BioSamples. Details are provided here.
The FAANG sample submission spreadsheet template provides a detailed questionnaire for each sample and hence our database system was modeled to accept the FAANG excel template. However, the required fields in the spreadsheet are the Animal and Specimen sheets; with the Animal-‘Sample Name’, Animal-‘Organism’, Specimen-‘Sample Name’ and Specimen-‘Organism Part’ column filled. Also, the Specimen-‘Derived from’ should be the same as the Animal-‘Sample Name’ column of each sample.
| Header | Status | Description |
|---|---|---|
| Sample name | required |
Sample identification number |
| Sample description | optional |
Description of sample |
| Derived from | required |
Animal identification number |
| Organism | required |
Species name |
| Organism part | required |
Name of tissue |
| First name | optional |
Person's first name |
| Middle initial | optional |
Person's middle initial |
| Last name | optional |
Person's last name |
| Organization | optional |
Institutional name |
To download the FAANG BioSamples template click here.
To download the tab-delimited template click here.
Otherwise, the sample information can be manually inserted using SQL insert statements.
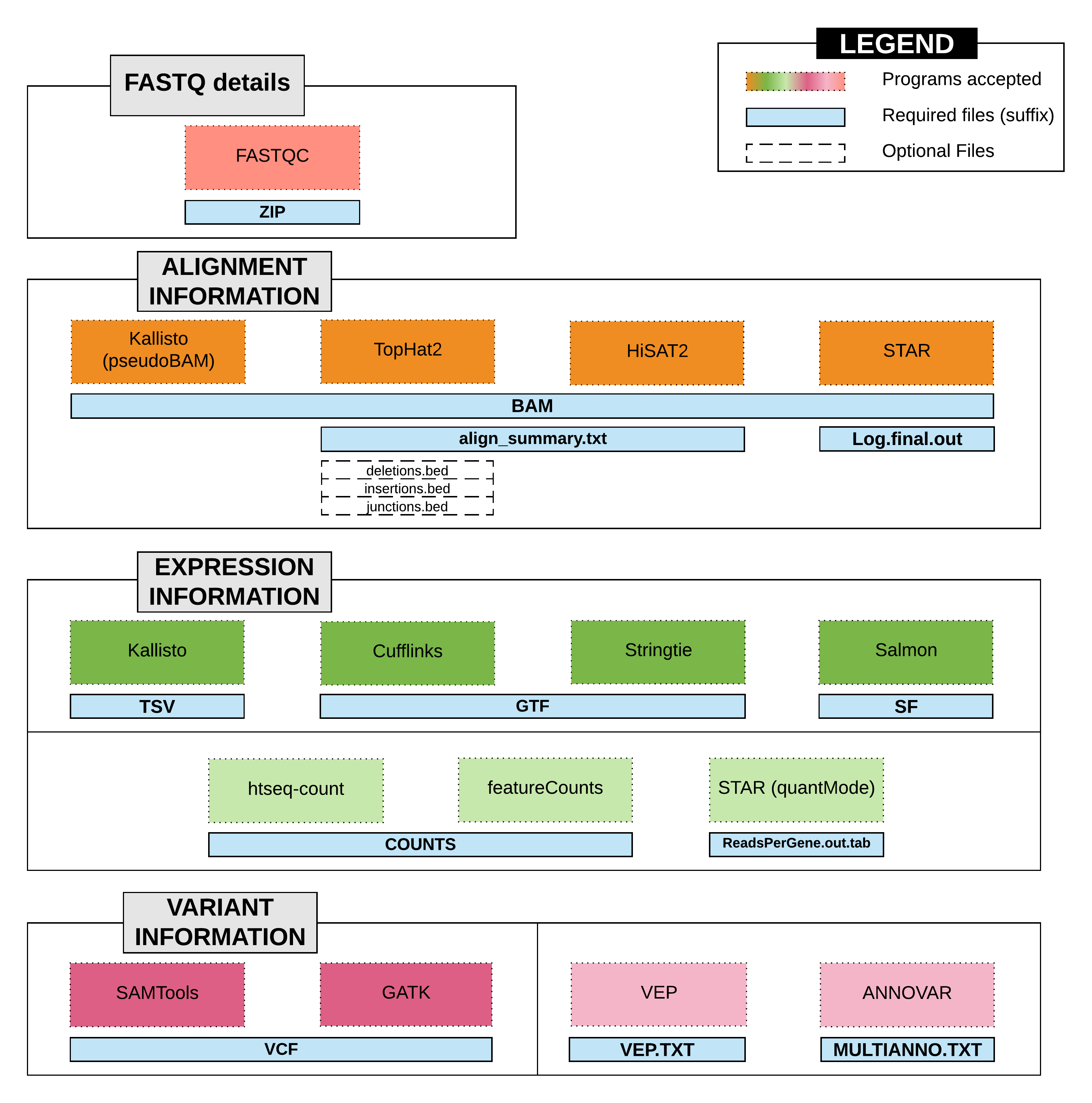
- Alignment Information. The Alignment information is comprised of the alignment files generated from either RNAseq read mappers. Depending on the RNAseq read mapper used, the expected files are;
- BAM file ( suffix: .bam )
- Summary file ( i.e. align_summary.txt or Log.final.out ) NOTE: HISAT2 alignment summary, produced as stardard output at default, must be saved as 'align_summary.txt' file
- Bed files ( i.e. deletions.bed , insertions.bed , junctions.bed ) NOTE: The bed files are optional.
- Expression Information. The Expression information consists of the genes FPKM (Fragments Per Kilobase of transcript per million) file generated using Cufflinks or StringTie, and genomic features read counts using various feature summarization tools like HTSeq-count and featurecount. The required files are;
- StringTie / Cufflinks
- Transcripts GTF ( suffix: .gtf ) NOTE: The transcripts details should be saved with the suffix '.gtf'
- Kallisto : Transcripts TSV file ( suffix: .tsv ), or
- Salmon : Transcripts SF file ( suffix: .sf ), or
- Feature summarization
- htseq-count / featureCounts ( suffix: .counts ) NOTE: The read counts should be saved with the suffix '.counts'
- STAR ( suffix: ReadsPerGene.out.tab )
- Variant Information. The Variant information includes the variant VCF file ( suffix: .vcf ) from variant callers, such as GATK, SAMtools (BCFtools).
- Ensembl Variant Effect Predictor (VEP) : The output file should be named with suffix .vep.txt.
- ANNOVAR : The default output file with suffix multianno.txt will be imported.
Optionally, the functional annotations of variants predicted by different bioinformatics tools can also be provided in a tab-delimited format. TransAtlasDB currently accepts functional annotations from two annotation software;
- FASTQ details. This is an optional feature to store the sequence files information if previously unavailable. This requires the FASTQC zipped folder (suffix: .zip ).
OUTPUT FORMAT
TransAtlasDB outputs user-defined queries as a tab-delimited table.
This table is the default output format which is accepted by most text editors or statistics tools such as Microsoft Excel, R and JMP software.
Asides from the tab-delimited format for exporting results, the variant information can be generated as a VCF output using the
Variant Call Format
Predicted functional annotations and sample metadata are added in the INFO field of the VCF file, using the key “CSQ” and “MTD” respectively.
Data fields are encoded separated by “|”; the order of fields is written in the VCF header.
VCFs produced by TransAtlasDB follow the standard VCF version 4 file format, and can be used for further downstream analysis or visualization using various variant viewers such as the University of California Santa Cruz (UCSC) Genome Browser, JBrowse, Integrative Genomics Viewer (IGV), and other programs that accept VCF files.
Please click the menu items to navigate through this repository. If you have questions, comments and bug reports, please email me directly.
Thank you very much for your help and support!
Back to Top